Somatic data
Somatic VCFs detected as somatic only (tumor minus normal) are analysed for mutational signatures
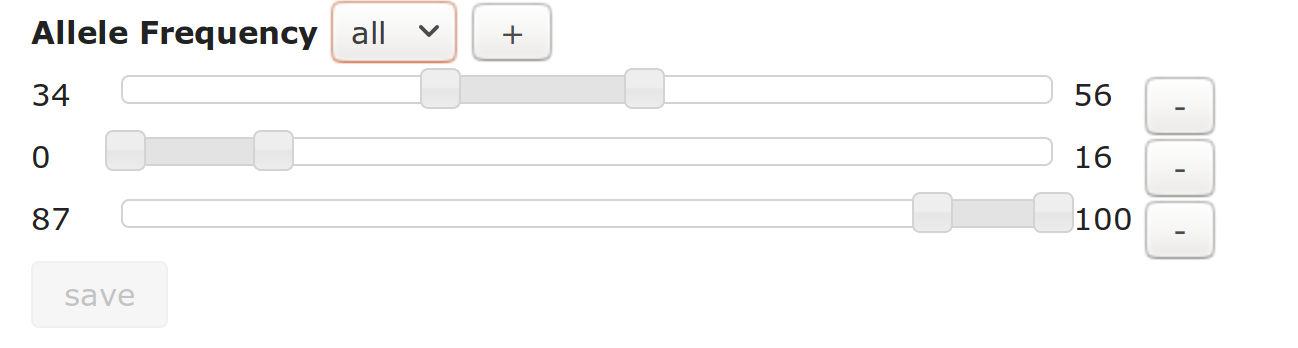
Allele Frequency
If the VCF contains an “AF” value, we will use that. Otherwise we will
We do not import the AF value from the VCF, but instead [normalize](../vcf_samples.md#VCF Normalization) the data then recalculate AF to be AD / sum(AD for all variants at locus)
In an analysis, nodes that represent one or more VCF samples (Sample, Cohort and Trio nodes) can filter by allele frequency.
Click the “+” button to add more sliders for AF ranges (between 0 and 100%) you will allow through (AF in any of the slider ranges will be allowed through)
For nodes with multiple sample (Cohort and Trio nodes):
all: all samples must have AF within the range sliders
any: at least one sample has AF within the range sliders